
Figure 2
Map of 10 sampling locations along a transect conducted in the Yolo Bypass, California on March 3, 2004.
Figure 3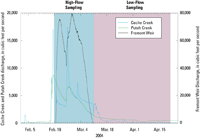
Discharge following a significant rainfall–runoff event at three inputs to the Yolo Bypass during 2004 high and low-flow sampling events.
Sample Collection and Analytical Methods
Sample Collection
Sites were selected on each of the major surface water inputs to the Yolo Bypass. Samples were collected from six sites: Sacramento River at Knights Landing, (abbreviated as Sacramento River), Sacramento Slough near Knights Landing (Sacramento Slough), Cache Creek Inflow to the Settling Basin near Woodland (Cache Creek), South Fork of Putah Creek at Mace Rd near Davis (Putah Creek), Knights Landing Ridge Cut near Knights Landing (KL Ridge Cut), and Willow Slough Bypass at County Rd 105 near Davis (Willow Slough) (table 2, fig. 1). All sites are located as close to the Bypass as feasible (fig. 1). Additional samples were collected from three sites located within the Bypass: Toe Drain at Yolo Bypass near West Sacramento (Toe Drain), Woodland R1 (Woodland), and Yolo Bypass Demonstration Pond (Yolo pond) (table 2, fig. 1), and nine stations along a transect across the Bypass with Toe Drain as the 10th station along the transect (table 2, fig. 2).
Table 2
(View this table on a separate page.)
Surface water, suspended and bed sediment sampling sites in the Yolo Bypass and its tributaries, California, and sample matrix collected.
[Horizontal Datum is NAD 83. dms, degrees, minutes, seconds; km, kilometer]
[Horizontal Datum is NAD 83. dms, degrees, minutes, seconds; km, kilometer]
| Site name | USGS site identification no. | Latitude (dms) | Longitude (dms) | Distance to bypass (km) | Sample matrix collected |
|---|---|---|---|---|---|
| Cache Creek Inflow to Settling Basin near Woodland | 384340121434401 | 38°43'40" | 121°43'48" | 9.2 | Water, bed sediment |
| Knights Landing Ridge Cut near Knights Landing | 384455121414001 | 38°44'55" | 121°41'40" | 3.9 | Water, suspended and bed sediment |
| Sacramento River at Knights Landing | 11391000 | 38°47'06" | 121°39'16" | 9.1 | Water |
| Sacramento Slough near Knights Landing | 11391100 | 38°48'11" | 121°42'59" | 2 | Water |
| South Fork Putah Creek at Mace Rd near Davis | 383109121414601 | 38°31 09" | 121°41'46" | 3.5 | Water, suspended and bed sediment |
| Toe Drain at Yolo Bypass near West Sacramento | 383425121350201 | 38°34'25" | 121°35'02" | Within bypass | Water and bed sediment |
| Willow Slough Bypass at County Rd 105 near Davis | 383524121403401 | 38°35'24" | 121°40'34" | 4.7 | Water, suspended and bed sediment |
| Woodland R1 | Within bypass | Bed sediment | |||
| Yolo Bypass Demonstration Pond | Within bypass | Water |
Primary field sampling began in mid-February of 2004 following a significant rainfall and runoff event in the area, and continued through the end of April 2004 (fig. 3). Water for the high-flow sampling was collected from February 19 to March 11 on a weekly basis from the six inputs to the Bypass. Surface water for the low-flow sampling was collected weekly between March 17 and April 21 from the two sites within the bypass (Yolo Pond and Toe Drain) and Putah Creek. On March 3, water was collected from 10 stations along a single transect across the width of the Bypass (fig. 2). Suspended-sediment samples were collected on February 20 from Putah Creek and KL Ridge Cut.
Bed-sediment samples were collected in September 2004 by personnel from a local environmental consulting firm (Larry Walker Associates, Davis, California). Samples were collected at four input sites (Cache Creek, KL Ridge Cut, Putah Creek, and Willow Slough) and two sites within the Bypass (Woodland and Toe Drain) (Larry Walker Associates, 2005).
In January 2005, water and suspended-sediment samples were collected once during a significant rainfall runoff event at the KL Ridge Cut and Willow Slough sites.
Water
Samples were collected for analysis of pesticides, suspended-sediment concentrations, and water-quality parameters (pH, specific conductance, and temperature) at all sites except Woodland. Water samples were collected as mid-channel grabs from bridges using a weighted, two-bottle sampler. Sample water was collected at a depth of approximately 0.5 m directly into one 1-L baked, glass bottle and one 500-mL glass milk bottle for pesticide and suspended-sediment concentration analyses, respectively. Samples were immediately placed on ice and transported to the Sacramento laboratory. Whole water samples collected for analysis of suspended-sediment concentration were shipped within 1 month of collection to the USGS’s Sediment Laboratory in Marina, California. Samples were also collected for pesticide analysis from nine stations and the Toe Drain site along one transect located immediately south of the Interstate 80 causeway within the Bypass (fig. 2). Water was collected by hand-dipping 1-L baked, amber glass bottles just below the water surface at 10 stations spaced equally across the Bypass. These samples were preserved on ice and transported to the Sacramento laboratory.
Suspended Sediment
Large volume water samples were collected at three sites (KL Ridge Cut, Putah Creek, and Willow Slough) and processed to isolate suspended sediments. At each site, approximately 200 L of water were collected using a peristaltic pump equipped with a stainless steel and Teflon inlet hose. Water was pumped at multiple stations across each channel profile, and at each station the inlet hose was suspended at multiple depths through the water column. All samples were collected and transported in 20-L stainless steel soda kegs to the Sacramento laboratory.
Bed Sediment
Bed-sediment samples were collected from four input sites and two sites within the Bypass (table 2). Sediment was collected from the top 2 centimeters of undisturbed stream bottom in areas of active deposition in 500-mL pre-cleaned glass jars. Samples were then shipped on ice to the Sacramento laboratory and stored frozen at –20 °C until analysis.
Analysis of Dissolved Pesticides
Water samples were filtered through baked 0.7 µm glass fiber filters within 24 hours of collection. Terbuthylazine was added to each sample as a recovery surrogate to provide quantitative data on extraction efficiency and the samples were extracted onto C8 solid phase extraction cartridges. The cartridges were dried using compressed carbon dioxide, frozen, and stored for up to 6 months at –20 °C. Prior to analysis, the cartridges were thawed, eluted using 9 mL of ethyl acetate, and concentrated for analysis. Deuterated polycyclic aromatic hydrocarbon (PAH) compounds were used as an internal standard and included d10-acenaphthene, d10-phenanthrene, and d10-pyrene. All extracts were analyzed for 27 pesticides using a Varian Saturn 2000 gas chromatograph mass spectrometer (GC/MS) with ion trap detection. Details of the analytical method are described in Crepeau and others (2000).
Analysis of Pesticides Associated with Bed and Suspended Sediments
Large volume water samples were processed to isolate suspended-sediment particles using a Westfalia continuous-flow centrifuge, within 6 hours of collection. During this process, sample water was passed through the centrifuge at a rate of 2 L/min using a peristaltic pump. This flow rate has been shown to be optimal for the collection of suspended-sediment particles (Horowitz and others, 1989). In addition, a single 1-L water sample was collected from the centrifuge effluent and analyzed for dissolved pesticides.
Following centrifugation, the concentrated sediment and sediment-water slurry were removed from the stainless steel centrifuge bowls and further dewatered by centrifuging for 20 minutes at 10,000 rpm using a high speed refrigerated centrifuge (Sorvall RC-5B centrifuge, DuPont Company, Wilmington, Delaware). The water separated from the samples during this step was decanted, and the remaining sediments were placed in precleaned glass jars and stored frozen until analysis.
The sediments were analyzed using the method described by LeBlanc and others (2004) with slight modifications to lower the MDL and limit matrix interference. The modified method, discussed in detail in this section, includes six compounds not analyzed in the original method and excludes three previously analyzed compounds. To avoid cross contamination and artifacts associated with drying and to increase extraction efficiency of the MASE, wet sediments were used (Jayaraman and others, 2001). Approximately 5 g of sediment (dry weight) were fortified with a labeled surrogate recovery solution containing 400-ng 13C-labeled trifluralin, chlorpyrifos, p,p′-DDE, and permethrin (cis/trans mixture) (Cambridge Isotope Laboratories Inc., Andover, Massachusetts). The amount of moisture in the sediment was set at 50 percent prior to MASE by adding between 0.2 and 1.5 mL of organic free deionized water depending on the moisture content of each sediment sample. The sediment samples were extracted two times with a mixture of dichloromethane (DCM) and acetone (50:50 v/v) using an MSP 1000 (CEM Corporation, Mathews, North Carolina). Details of the MASE procedure are described in Leblanc and others (2004).
Following extraction, the samples were decanted through glass funnels packed with approximately 30 g of sodium sulfate to remove excess water. Extracts were reduced at 25 °C and 0.6 atm to 0.75 mL using a Turbovap II (Zymark Corporation, Hopkinton, Maryland). Sediment matrix was removed by passing the sample extract via vertical flow under gravity through two stacked solid-phase extraction (SPE) cartridges containing different sorbents. A 6 mL, 500 mg, nonporous, graphitized carbon SPE (Restek Corporation, Bellefonte, Virginia) was stacked on top of a 500 mg Alumina SPE (Varian Inc., Palo Alto, California) and washed with 10 mL of DCM to remove cartridge impurities. The organic rich, colored sample extract was added to the cartridges, rinsed in tandem with 10 mL of DCM, and collected as fraction 1 (F1). The carbon SPE was removed and the Alumina SPE was eluted with 10 mL of ethyl acetate and DCM (50:50 v/v) and collected as fraction 2 (F2). With the exception of the triazines/triazinones, carbamates, and napropamide, most pesticide classes were eluted primarily in the F1 with minimal carryover into the F2. Only molinate and methidathion split between the F1 and F2. Because the F2 contained more sample matrix than the F1, the two fractions were analyzed separately to reduce interferences with the pyrethroids in the first fraction and improve the (MDLs). Concentrations of each analyte were calculated in each fraction separately and then summed together to give a final reported value.
Both fractions were evaporated separately under a gentle stream of highly purified nitrogen gas (N-evap, Organomation Associates, Berlin, Massachusetts) to 0.5 mL and exchanged into ethyl acetate. Sulfur, found only in the F1 extracts, was removed using a gel permeation/high pressure liquid chromatography (GPC/HPLC) system. The F1 and F2 extracts were reduced under a gentle stream of N2 to 0.2 mL, and 40 µL of the deuterated internal PAH standard was added. The extracts were analyzed by GC/MS for a suite of 41 pesticides.
Five pyrethroids were added to the method, which included deltamethrin, fenpropathrin, phenothrin, resmethrin, and tau-fluvalinate (table 3). Diethylatyl-ethyl and azinphos-methyl were the two compounds removed from the original method developed by LeBlanc and others (2004).
Table 3
(View this table on a separate page.)
Mean recovery of pesticides from matrix spikes (n = 9) using various bed (n = 5) and suspended (n = 4) sediment and method detection limits (MDL).
[All MDLs were determined in Cache Creek sediment only (n = 7). µg/kg, microgram per kilogram; SD, standard deviation; MDL, method detection limit; NA, not available]
[All MDLs were determined in Cache Creek sediment only (n = 7). µg/kg, microgram per kilogram; SD, standard deviation; MDL, method detection limit; NA, not available]
| Mean ± SD (%) | MDL (ng/g) | |
|---|---|---|
| Triazines/Triazones | ||
| Atrazine¹ | 85.0 ± 5.1 | 1.7 |
| Hexazinone | 114 ± 12.7 | 2.3 |
| Prometryn¹ | 89.3 ± 9.0 | 1.9 |
| Simazine¹ | 91.3 ± 7.8 | 1.4 |
| Anilines | ||
| Ethalfluralin¹ | 82.8 ± 8.9 | 1.2 |
| Pendamethalin | 105 ± 5.1 | 1.5 |
| Trifluralin | 88.9 ± 13.6 | 1.1 |
| Chloacetanilides | ||
| Alachlor¹ | 86.1 ± 10.9 | 1.4 |
| Metolachlor¹ | 85.1 ± 5.6 | 1.7 |
| Carbamates | ||
| Carbaryl | 103 ± 8.0 | 2.2 |
| Carbofuran¹ | 102 ± 13.0 | 5.3 |
| Thiocarbamates | ||
| Butylate¹ | 60.0 ± 6.6 | 1.1 |
| Cycloate¹ | 66.6 ± 5.5 | 0.8 |
| EPTC¹ | 62.3 ± 8.1 | 1.4 |
| Molinate | 62.9 ± 7.7 | 0.6 |
| Pebulate¹ | 60.6 ± 6.9 | 0.9 |
| Thiobencarb | 93.9 ± 10.7 | 1.6 |
| Organochlorines | ||
| p,p'-DDD | 85.4 ± 12.7 | 1.3 |
| p,p′-DDE | 85.1 ± 9.1 | 1.5 |
| p,p′-DDT | 85.2 ± 12.4 | 1.9 |
| Organophosphates | ||
| Chlorpyrifos | 82.5 ± 6.0 | 0.8 |
| Diazinon¹ | 85.1 ± 14.8 | 0.6 |
| Malathion¹ | 94.4 ± 7.8 | 2.2 |
| Methidathion¹ | 102 ± 10.5 | 1.5 |
| Methylparathion¹ | 103 ± 7.4 | 2 |
| Phosmet¹ | 93.8 ± 17.0 | 2.4 |
| Pyrethroids | ||
| Bifenthrin | 84.2 ± 15.1 | 2.3 |
| Cyfluthrin¹ | 81.5 ± 7.3 | 7.9 |
| Cypermethrin¹ | 86.0 ± 14.1 | 5.6 |
| Deltamethrin¹ | 88.5 ± 15.1 | 1.1 |
| Esfevalerate¹ | 80.1 ± 6.6 | 1.8 |
| Fenpropathrin¹ | 85.3 ± 17.9 | 1.4 |
| Lambda-Cyhalothrin¹ | 77.1 ± 9.8 | 1.6 |
| Permethrin¹ | 79.0 ± 12.8 | 1.2 |
| Sumithrin¹ | 92.8 ± 16.3 | 2.9 |
| Tau-fluvalinate | 83.5 ± 14.9 | 1.1 |
| Miscellaneous | ||
| DCPA | 82.4 ± 8.4 | 1.5 |
| Napropamide | 98.2 ± 8.5 | 1.6 |
| Oxuflurofen | 92.7 ± 10.5 | 2.5 |
| Piperonyl butoxide¹ | 106 ± 10.0 | 1.3 |
Quality Assurance and Quality Control
Dissolved pesticide concentrations were validated against a comprehensive set of quality control parameters including laboratory and field blanks, matrix spikes, replicate samples, and surrogate recovery. Laboratory and field blanks were analyzed every 10–20 samples for a total of 6 in 2004 and 1 in 2005. No pesticides were detected in any of the blanks. Replicate samples (6) were analyzed constituting approximately 10 percent of the samples and were within 25 percent agreement for all pesticides detected. Matrix spikes were analyzed in approximately 10 percent of the samples as part of the described method validation with recoveries ranging from 80 to 120 percent. Terbuthylazine was used as a recovery surrogate to assess the efficiency of sample extraction. The average percentage recovery and standard deviation of the recovery surrogate were calculated for each site. Sample data were excluded if the recovery of terbuthylazine was outside the mean plus or minus two standard deviations.
Sediment matrix spikes, method blanks, and replicate samples were also processed for quality control purposes. During final method testing, 200 ng of each pesticide listed in table 3 was spiked into five bed sediment and four suspended-sediment samples. Matrix spike percentage recoveries ranged from 60 to 114 percent (table 3). Replicate samples constituted approximately 10 percent of the total samples analyzed, and the differences between replicates were less than 25 percent for all pesticides detected. No pesticides were detected in any blank sample run with each batch of five sediment samples. Recovery of the sediment surrogate mixture was used to monitor the efficiency of each extraction. The average percentage recoveries of 13C-labeled trifluralin, chlorpyrifos, p,p′-DDE, and permethrin (cis/trans mixture) were 92.0 ± 10.2, 93.0 ± 9.0, 87.5 ± 13.3 and 95.2 ± 10.0, respectively.
Method Detection Limits
Surface water method detection limits were validated in a previous study (Orlando and others, 2004) using the EPA procedure (U.S. Environmental Protection Agency, 1992; table 4). Water used for the MDLs was collected in 2001 and 2002 from the San Joaquin River near Vernalis (USGS site ID number 11303500), which has similar water chemistry to the sites sampled in the study.
Table 4
(View this table on a separate page.)
Method detection limits for pesticides analyzed in surface water in 2001 and 2002.
[The method detection limits for pesticides analyzed in surface water in 2001 and 2002 are taken from Orlando and others, 2004. Method detection limits not available for Bifenthrin, Cyfluthrin, Cypermethrin, Deltamethrin, Esfenvalerate, Fenpropathrin, Lambda-cyhalothrin, Resmethrin, Permethrin, Phenothrin, p,p′-DDD, p,p′-DDE, p,p′-DDT, Tau-fluvalinate; ng/L, nanogram per liter]
[The method detection limits for pesticides analyzed in surface water in 2001 and 2002 are taken from Orlando and others, 2004. Method detection limits not available for Bifenthrin, Cyfluthrin, Cypermethrin, Deltamethrin, Esfenvalerate, Fenpropathrin, Lambda-cyhalothrin, Resmethrin, Permethrin, Phenothrin, p,p′-DDD, p,p′-DDE, p,p′-DDT, Tau-fluvalinate; ng/L, nanogram per liter]
| Pesticide | Method detection limit (ng/L) |
|---|---|
| Alachlor¹ | 2.1 |
| Atrazine¹ | 4.2 |
| Butylate¹ | 1.8 |
| Carbaryl | 4.2 |
| Carbofuran¹ | 3.3 |
| Chlorpyrifos¹ | 4.2 |
| Cycloate¹ | 1.5 |
| DCPA¹ | 1.2 |
| Diazinon | 3.6 |
| EPTC | 4.5 |
| Ethalfluralin¹ | 2.4 |
| Hexazinone | 5.7 |
| Malathion¹ | 2.1 |
| Methidathion | 5.4 |
| Methyl parathion¹ | 4.2 |
| Metolachlor | 3.3 |
| Molinate | 2.7 |
| Napropamide | 7.2 |
| Oxyfluorfen | 4.2 |
| Pebulate¹ | 0.6 |
| Pendimethalin | 2.4 |
| Phosmet¹ | 4.2 |
| Piperonyl butoxide¹ | 3.3 |
| Prometryn¹ | 2.7 |
| Simazine | 6.9 |
| Thiobencarb | 3.9 |
| Trifluralin | 3.0 |
MDLs for the sediment samples were determined using seven replicates of Cache Creek sediment collected for this study. Cache Creek was chosen because it had low background pesticide concentrations and was similar in organic carbon content to the other sites. A mixture containing approximately 50 ng of each analyte (approximately10 ng/g dry weight) was added to the sediment and carried through the entire procedure. The method detection limits for each compound in sediment and water are listed in tables 3and 4.
The MDL was calculated for each pesticide using the following equation:
MDL = S × t (n–1, 1–α = 0.99)
where:
MDL = method detection limit (µg/kg)
S = standard deviation of replicate samples
n = number of replicates
t = value of Student’s t statistic at 6 degrees of freedom and 99 percent confidence level.
MDLs for sediment ranged from 0.6 to 7.9 µg/kg (table 3), whereas MDLs for dissolved pesticides ranged from 0.6 to 7.2 ng/L (table 4). Analytes can be identified at concentrations less than the MDL with a lower confidence in the actual value; therefore, these concentrations are reported as estimated values.
Sediment Organic Carbon Analysis
Suspended and bed sediments were analyzed for organic carbon content using a Perkin Elmer CHNS/O analyzer (Perkin Elmer Corporation, Norwalk, Connecticut). Sediments were combusted at 925 °C in silver boats after being exposed to concentrated HCl fumes in a desiccator for 24 hours to remove inorganic carbon. Before analysis, sediments were dried to a constant weight at 100 °C for 3 hours. Acetanilimide was used for instrument calibration of elemental carbon and nitrogen.
Analysis of Suspended Sediments and Water-Quality Parameters
Whole water samples were analyzed for suspended-sediment concentration at the U.S. Geological Survey Sediment Laboratory in Marina, California. Details of the analytical method can be found in Guy (1969). Analytical results of single-blind quality control samples provided by the USGS’s Sediment Laboratory Quality Assurance Project show that laboratory performance during the period of this study was satisfactory (U.S. Geological Survey, 2005).
Water parameters (pH, specific conductance, and water temperature) were measured in whole-water samples on site or at the Sacramento laboratory within 24 hours of sample collection. Specific conductance and pH were measured using two handheld instruments, (Cole Parmer Model 141-61 and Orion Model 250A, respectively), following methods described in the USGS’s National Field Manual (Wilde and Radtke, 1998). Water temperature was measured in the field at the time of collection using a digital thermometer.